Мукополисахаридоз
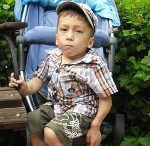
Мукополисахаридозы – это группа генетически обусловленных заболеваний, возникающих вследствие нарушения обмена кислых мукополисахаридов (гликозаминогликанов). Характерны системные поражения скелета и задержка физического развития. При некоторых формах наблюдается умственная отсталость. Возможны нарушения сердечной деятельности, патология органов зрения, образование грыж, неврологические нарушения, гипертрихоз, увеличение печени и селезенки. Диагноз выставляется на основании клинических признаков, данных рентгенографии и других исследований. Лечение симптоматическое.
МКБ-10
E76.0 E76.1 E76.2
- Виды мукополисахаридоза
- Мукополисахаридоз типа IH
- Мукополисахаридоз типа I-S
- Мукополисахаридоз типа II
- Мукополисахаридоз типа III
- Мукополисахаридоз типа IV
- Другие типы мукополисахаридоза
Общие сведения
Мукополисахаридозы – группа генетических заболеваний, сопровождающихся накоплением кислых мукополисахаридов в органах и тканях. Причиной развития является передающаяся по наследству неполноценность лизосомных ферментов. Впервые мукополисахаридоз был описан Гурлер в 1917 году. Лечение мукополисахаридоза осуществляют травматологи-ортопеды при участии кардиологов, офтальмологов, неврологов, отоларингологов и других специалистов.
Виды мукополисахаридоза
Мукополисахаридоз типа IH
Мукополисахаридоз типа IH или синдром Гурлер встречается у 1 новорожденного из 20-25 тысяч. Симптомы мукополисахаридоза появляются в течение первого года жизни, полная клиническая картина формируется к 1-2 годам. Для данной формы мукополисахаридоза характерны грубые черты лица и деформация черепа в форме лодочного киля (скароцефалия). Из-за увеличенных аденоидов и пороков развития в области лица и носа больные дышат ртом. Отмечаются прогрессирующие деформации конечностей и других частей скелета, отставание в росте.
Позвоночник пациентов с мукополисахаридозом искривлен, из-за чего в положении сидя возникает симптом «кошачьей спины». Отмечается укорочение шеи, высокое расположение лопаток и выстояние нижних ребер. Кисти широкие, напоминающие когтистую лапу. Со временем у больных мукополисахаридозом формируются контрактуры суставов. Вначале поражаются локтевые и плечевые суставы, затем – коленные, тазобедренные и голеностопные. Из-за ограничения движений возникает характерная походка – на цыпочках с полусогнутыми ногами.
Тоны сердца приглушены, границы расширены. При аускультации определяются систолические шумы, на ЭКГ выявляется диффузное поражение миокарда. Передняя брюшная стенка пациентов с мукополисахаридозом ослаблена, живот увеличен, часто выявляются гидроцеле, пупочные и паховые грыжи. Печень и селезенка увеличены. Характерны нарушения зрения и слуха. Возможно помутнение и увеличение роговицы, пигментная дистрофия сетчатки, глаукома, атрофия зрительных нервов и застойные явления в области глазного дна. У больных мукополисахаридозом этого типа развивается прогрессирующая умственная отсталость. Могут возникать нарушения координации движений, парезы и параличи. Выявляется избыточный рост пушковых волос.
Рентгенография позвоночника пациентов с мукополисахаридозом свидетельствует о характерной деформации позвонков. Позвонки имеют кубовидную форму, их контуры закруглены, в переходном отделе выявляется скошенность передневерхних углов, углообразный кифоз, утолщение и укорочение отростков. На рентгенографии грудной клетки определяется утолщение передних и истончение задних отделов ребер, сопровождающиеся их лопатовидной или саблевидной деформацией, укорочение и деформация ключиц, уменьшение и смещение головок плечевых костей.
Рентгенография таза подтверждает сужение тазового кольца, скошенность вертлужных впадин и уменьшение головок бедренных костей. На рентгенограммах трубчатых костей выявляется истончение кортикального слоя и расширение костномозгового канала. Рентгенография костей кисти свидетельствует о недоразвитии ногтевых фаланг, укорочении и расширении пястных костей, проксимальных и средних фаланг. На рентгенограммах черепа определяется недоразвитие костей лицевого черепа, краниостеноз и макроцефалия.
Мукополисахаридоз типа I-S
Мукополисахаридоз типа I-S или болезнь Шейе (описана в 1962 г. американским офтальмологом Шейе) является более поздним, относительно благоприятно протекающим вариантом мукополисахаридоза типа IH (синдрома Гурлер). До 3-6 лет развитие детей соответствует норме. Первым признаком мукополисахаридоза становятся сгибательные контрактуры пальцев рук. В последующем ограничивается разгибания в лучезапястных, локтевых и плечевых суставах. Контрактуры нижних конечностей, как правило, слабо выражены. Полная клиническая картина мукополисахаридоза формируется к началу подросткового возраста.
Пациенты с мукополисахаридозом коренастые, невысокие, с грубыми чертами лица и хорошо развитой мускулатурой. Отмечается повышенное оволосение (гипертрихоз). Часто возникают паховые или пупочные грыжи. Кожа на пальцах натянута и утолщена. Из-за сдавления срединного нерва возможно развитие синдрома запястного канала, сопровождающееся атрофией мышц области тенара и парестезиями в области III-IV пальцев. У некоторых больных мукополисахаридозом выявляется аортальный стеноз, недостаточность клапанов аорты, пигментная дистрофия сетчатки, глаукома и помутнение роговицы. Интеллект в норме, увеличение селезенки и печени нехарактерно. На рентгенограммах определяется картина, аналогичная мукополисахаридозу типа IH, но патологические изменения выражены менее резко.
Мукополисахаридоз типа II
Мукополисахаридоз типа II или синдром Хантера чаще выявляется у мальчиков и обычно развивается на 2-3 году жизни. Как и при мукополисахаридозе типа IH наблюдается скафоцефалия, огрубление черт лица, низкий голос и затруднения дыхания из-за деформации лицевого скелета. При этом характерный для мукополисахаридоза типа IH кифоз обычно не выявляется, симптом «кошачьей спины» отрицательный. Пациенты часто страдают от респираторных инфекций (бронхитов, трахеитов, пневмоний). Постепенно развиваются нарушения координации движений, возрастает агрессивность. Настроение неустойчивое, с резкими перепадами.
Также при данной форме мукополисахаридоза наблюдается незначительное увеличение селезенки и печени, прогрессирующая тугоухость, узелки на коже спины и некоторое снижение интеллекта. В последующем может развиться помутнение роговицы. Рентгенологическая картина – как при мукополисахаридозе типа I-S. Возможны два варианта развития болезни: благоприятный (вариант В) и неблагоприятный (вариант А). При благоприятном варианте симптомы выражены нерезко, иногда возникает незначительная умственная отсталость, пациенты с мукополисахаридозом могут доживать до 30 и более лет. Для неблагоприятного варианта характерна яркая клиническая картина и серьезные нарушения интеллекта. Летальный исход наступает в подростковом возрасте.
Мукополисахаридоз типа III
Мукополисахаридоз типа III или болезнь Санфилиппо (описана в 1963 г. американским педиатром Санфилиппо) развивается у 1 новорожденного из 100-200 тысяч. В первые годы жизни развитие соответствует возрасту, иногда наблюдаются затруднения глотания и неуклюжая походка. В возрасте 3-5 лет ребенок с мукополисахаридозом становится апатичным и начинает отставать в развитии. Возникают нарушения речи, огрубление черт лица, недержание мочи и кала. Со временем умственная отсталость прогрессирует и занимает центральное положение в клинической картине этого типа мукополисахаридоза.
Наряду с нарушениями интеллекта наблюдается умеренное увеличение печени и селезенки, повышенное оволосение, контрактуры и задержка роста. Патология со стороны глаз и сердечно-сосудистой системы для этой формы мукополисахаридоза нехарактерна. Рентгенологическая картина – без изменений или как при мукополисахаридозе типа I-S, но менее выраженная. Больные мукополисахаридозом третьего типа, как правило, погибают в возрасте 10-20 лет от инфекционных осложнений.
Мукополисахаридоз типа IV
Мукополисахаридоз типа IV или болезнь Моркио (описана в 1929 г. уругвайским педиатром Моркио) наблюдается у 1 новорожденного из 40 тысяч. До 1-3 лет дети развиваются нормально. В последующем возникает значительное отставание в росте, укорочение шеи и туловища, контрактуры и вальгусная деформация конечностей, сколиоз или кифоз, разнообразные деформации грудной клетки, снижение силы мышц, утолщение кожи и огрубление черт лица. При этом типе мукополисахаридоза часто развиваются паховые и пупочные грыжи, дистрофия роговицы и тугоухость. Интеллект сохранен.
На рентгенограммах позвоночника определяется кифоз, сколиоз, расширение и уплощение тел позвонков. При проведении рентгенографии таза и конечностей выявляются множественные деформации, неровность контуров, уплощение головок бедренных костей, укорочение костей предплечья и деформации стоп. Средняя продолжительность жизни больных мукополисахаридозом – менее 20 лет. Смерть при данной форме мукополисахаридоза наступает из-за сопутствующих заболеваний, осложняющихся сердечно-легочной недостаточностью.
Другие типы мукополисахаридоза
Мукополисахаридоз типа VI или болезнь Марото-Лами (описана в 1960 г. французами Лами и Марото) развивается в возрасте 2 года и старше. Возникает огрубление черт лица, отставание в росте, укорочение шеи, контрактуры суставов и бочкообразная деформация грудной клетки. Характерны частые простуды. Возможны грыжи, увеличение печени и селезенки. Интеллект не страдает. Наблюдается два варианта течения: классический и мукополисахаридоз со слабой выраженностью клинических проявлений. На рентгенограммах больных мукополисахаридозом определяется кубовидная или клиновидная деформация позвонков, треугольная деформация таза, недоразвитие и деформация головок бедренных костей, укорочение малоберцовых костей.
Мукополисахаридоз типа VII протекает, как мупоколисахаридоз типа III, различия выявляются только при проведении биохимических исследований. Мукополисахаридоз типа VIII по симптомам напоминает мукополисахаридоз типа IV, но, в отличие от него, сопровождается задержкой умственного развития.
Диагностика
Диагноз мукополисахаридоза устанавливается на основании характерной клинической и рентгенологической картины, выявления гликозаминогликанов в моче, изучения активности ферментов в клеточных культурах, генетического секвенирования. В ходе обследования больным мукополисахаридозом назначаются консультации различных специалистов: кардиолога, гастроэнтеролога, офтальмолога, отоларинголога, невролога, психиатра и т. д., проводятся инструментальные исследования для оценки состояния различных органов и систем.
Лечение мукополисахаридоза
Патогенетическая терапия не разработана. Лечение симптоматическое, может быть как консервативным, так и оперативным. Осуществляется профилактика и лечение респираторных инфекций, проводится коррекция нарушений зрения и слуха. При необходимости выполняются грыжесечение и герниопластика, операции по устранению контрактур и коррекции деформаций скелета.
Прогноз и профилактика
Прогноз при всех типах мукополисахаридоза неблагоприятный – продолжающееся накопление продуктов обмена в тканях приводит к усугублению патологических изменений со стороны всех органов и систем. Использование любых лечебных средств (переливания крови, введение гормонов и т. д.) при мукополисахаридозе обеспечивает лишь временное улучшение. Рекомендуется пренатальная профилактика.
Литература
1. Руководство по педиатрии. Врожденные и наследственные заболевания/ Новиков П.В., Баранов А.А., Каганов Б.С., Шиляев Р.Р — 2007
Мукополисахаридоз — симптомы и лечение
Что такое мукополисахаридоз? Причины возникновения, диагностику и методы лечения разберем в статье доктора Боровиковой Ольги Игоревны, генетика со стажем в 8 лет.
Над статьей доктора Боровиковой Ольги Игоревны работали литературный редактор Маргарита Тихонова , научный редактор Сергей Федосов и шеф-редактор Лада Родчанина

Генетик Cтаж — 8 лет
«Evi Clinic» («Эви Клиник»)
Клиника «OXY-center»
Медицинский центр «Андромед»
Дата публикации 24 декабря 2019 Обновлено 13 июня 2023Определение болезни. Причины заболевания
Мукополисахаридозы — это группа редких наследственных заболеваний соединительной ткани, связанных с нарушением обмена веществ. Они обусловлены нехваткой определённых ферментов, которые участвуют в переработке (расщеплении) гликозаминогликанов — сложных молекул сахара. В связи с чем эти молекулы скапливаются в организме человека в опасно большом количестве и приводят к различным изменениям [1] [14] .
Самые явные проявления мукополисахаридозов — множественные деформации костей и суставов и нарушение физического развития (задержка и утеря ранее приобретённых навыков. При определённых типах заболевания (синдромах Шейе, Гурлер — Шейе, Хантера, Санфилиппо — I, II, III типах мукополисахаридозов) помимо прочего нарушается умственное развитие, начиная с лёгких когнитивных нарушений и заканчивая глубокой деменцией [13] .
Изменения при мукополисахаридозах возникают в результате дефекта ферментного расщепления углеводной части молекулы мукополисахаридов (гликозаминогликанов). При этом в фибробластах и мезенхимальных клетках, которые способны трансформироваться в хрящевые, костные либо жировые клетки, накапливается хондроитинсульфат — вещество, являющееся основой хрящей. Это ведёт к нарушению структуры соединительной, костной и хрящевой ткани [13] .
Мукополисахаридозы встречаются очень редко: примерно у одного ребёнка среди 56-325 тысяч новорождённых. Их причиной является мутация . Эти болезни довольно сложно диагностировать из-за малой осведомлённости врачей о них. Поэтому больным часто выставляют другие диагнозы и проводят неадекватное лечение.
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением — это опасно для вашего здоровья!
Симптомы мукополисахаридоза
Мукополисахаридозы делятся на несколько типов. Они различаются первичным генным дефектом, патологическим ферментом, преимущественным поражением той или иной системы органов и тканей, возрастом начала заболевания и тяжестью его течения (см. классификацию ). В целом при этих генетических патологиях встречаются множественные нарушения: поражаются костная система, хрящи, печень, селезёнка, головной мозг, роговица глаза, органы лимфатической и дыхательной системы. Из-за особенностей строения дыхательных путей возникают частые инфекционные заболевания органов дыхания и слуха, что приводит к развитию тугоухости и респираторным расстройствам — бронхитам, пневмониям и др.
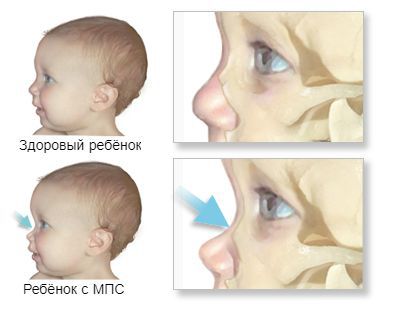
К самым частым проявлениям мукополисахаридозов относят низкорослость, волосатость и нарушения развития. Они дают о себе знать с раннего детства. Лицо ребёнка, как правило, приобретает грубые черты, становится «взрослым», а голова выглядит довольно большой из-за широкого лба и короткого носа. Губы и язык тоже становятся больше, чем у сверстников. Суставы пальцев плохо гнутся и двигаются. При отдельных типах заболевания нарушается слух и зрение, повреждаются сердечные клапаны и артерии [14] .
Патогенез мукополисахаридоза
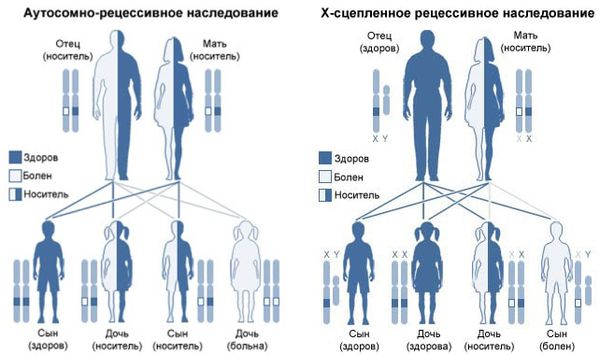
Все формы мукополисахаридоза наследуются по аутосомно-рецессивному типу, т. е. мутировавший ген должен быть у обоих родителей. Исключением является мукополисахаридоз II типа: он наследуется по Х-сцепленному рецессивному типу.
Каждая форма мукополисахаридоза отличается нарушением выработки различных ферментов, что приводит к изменению функции фермента, его нехватке или отсутствию. В результате этого нарушается ферментозависимый распад гликозаминогликанов: из-за нехватки определённых ферментов организм не в состоянии преобразовывать и сохранять эти полисахариды в тканях. Нерасщеплённые гликозаминогликаны через кровь распространяются по всему организму, что приводит к их избыточному накоплению в различных органах и системах [14] . Чаще всего эти вещества скапливаются в соединительной ткани, сердце, печени, селезёнке, нервной ткани и оболочках мозга:
- Отложение гликозаминогликанов в оболочках мозга способствует сужению субарахноидальных пространств, которые находятся под паутинной оболочкой головного мозга, и формированию гидроцефалии .
- Поражение нервных клеток приводит к задержке умственного развития, прогрессирующей деменции.
- Деформация метафизов (отделов трубчатых костей), утолщение межсуставных хрящей и суставных связок становятся причинами нарушения подвижности в суставах.
- В результате отложения гликозаминогликанов в миндалинах, трахее и голосовых связках возникает отёчность верхних дыхательных путей, нарушается дыхание, что приводит к нарушению вентиляции, частым респираторным заболеваниям, отитам, обструктивным состояниям (непроходимости дыхательных путей).
- Отложение гликозаминогликанов в тканях сердца сопровождается кардиомиопатиями и формированием клапанных пороков сердца.
- Повреждение канальцев почек может приводить к прогрессирующей артериальной гипертензии.
Классификация и стадии развития мукополисахаридоза
В зависимости от первичного генетического дефекта выделяют несколько типов мукополисахаридоза:
- МПС I типа включает в себя несколько подтипов: синдром Гурлер (H), Шейе (S) и Гурлер — Шейе (H/S) [6][7] . Мутация при данных фенотипах обнаруживается в гене IDUA, вызывает дефицит фермента альфа-L-идуронидазы [4] .
- МПС II типа — синдром Хантера [8] . Мутация при данном заболевании обнаруживается в гене IDS, вызывает дефицит либо отсутствие идуронат-2-сульфатазы, либо дефицит или отсутствие сульфоидуронат сульфатазы [5] .
- МПС III типа — синдром Санфилиппо — и меет несколько подтипов:
- МПС III А обусловлен мутацией в гене SGSH, приводит к нехватке гепаран-N-сульфатазы [9] ;
- МПС III В обусловлен мутацией в гене NAGLU, вызывает дефицит N-ацетил-α-D-глюкозаминидазы [10] ;
- МПС III С обусловлен мутацией в гене HGSNAT, вызывает дефицит гепаран-α-глюкозаминид-N-ацетилтрансферазы [11] ;
- МПС III D обусловлен мутацией в гене GNS, вызывает дефицит N-ацетилглюкозамин-6-сульфатазы [2][12] .
- МПС IV типа — синдром Моркио — делится на два подтипа:
- МПС IV А связан с мутацией в гене GALNS, вызывает дефицит галактозамин-6-сульфатазы;
- МПС IV В связан с мутацией в гене GLB1, вызывает недостаточность β-галактозидазы.
- МПС VI типа — синдром Марото — Лами — связан с мутацией гена ARSB, вызывает дефицит N-ацетилгалактозамин-4-сульфатазы [3] .
- МПС VII типа — синдром Слая — возникает вследствие мутации гена GUSB, приводит к дефициту β-глюкуронидазы.
- МПС IX типа является проявлением мутации гена HYAL1, приводит к недостаточности гиалуронидазы.
Остановимся подробнее на каждом типе мукополисахаридоза.
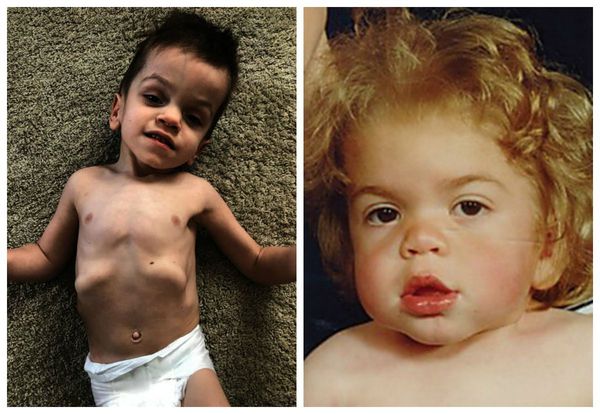
Мукополисахаридоз I H (синдром Гурлер). Основными симптомами заболевания являются: задержка психического и физического развития, умственная отсталость, грубые черты лица, пороки клапанов сердца, помутнение роговицы, низкорослость, тугоподвижность суставов. Первые признаки такого мукополисахаридоза появляются в течение первых 12 месяцев жизни младенца.
Иногда уже с самого рождения отмечается увеличение печени и селезёнки (гепатомегалия), пупочные или пахово-мошоночные грыжи. Ближе к 6-12 месяцам лицо приобретает грубые черты, напоминающие гаргулью: голова становится больше, выступают лобные бугры, появляются широкие скулы, уплощается и втягивается переносица, укорачиваются носовые ходы, изменяется форма носа, ноздри выворачиваются, рот постоянно полуоткрыт, увеличивается язык, губы становятся пухлыми. Низкорослость становится заметной ближе к 2-5 годам, рост обычно ниже 100 см. Пропорции тела нарушены, шея укорочена [3] .
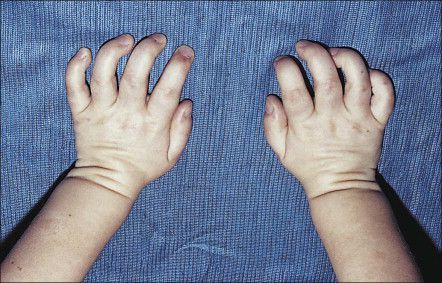
Очень часто у таких пациентов уменьшается подвижность крупных и мелких суставов, особенно пальцев, укорачиваются трубчатые кости, кисти рук деформируются и приобретают форму «лапы с когтями» [1] . Уменьшаются головки бедренных костей, из-за чего формируется дисплазия тазобедренных суставов. Подвздошные кости приобретают треугольную форму.
С развитием болезни к клинической картине присоединяются признаки поражения внутренних органов, сердца и сосудов, головного мозга, нервной системы.
Со стороны сердечно-сосудистой системы утолщаются клапаны, уменьшается диаметр артерий, нарушается сократимость сердечной мышцы, возникают кардиомиопатии, повышается артериальное давление. В итоге развивается сердечная недостаточность .
Основными проявлениями вовлечения нервной системы являются прогрессирующее снижение умственных способностей, задержка лингвистического (языкового) развития, гипотония мышц, снижение сухожильных рефлексов, патология черепно-мозговых нервов, снижение слуха [6] . Психическое и моторное развитие запаздывает, достигает максимального развития на уровне 2-4 лет, после чего останавливается и регрессирует, иногда переходя в полную деменцию. Ухудшает ситуацию прогрессирующая гидроцефалия [1] .
Поведение пациентов с синдромом Гурлер страдает из-за когнитивных нарушений, прогрессирующей тугоухости, бессонницы, связанной с ночной обструктивной задержкой дыхания . Со временем ребёнок становится всё активнее, развивается синдром дефицита внимания , агрессия, расстройства аутистического спектра.
При деформации позвоночника, утолщении оболочек спинного мозга изменяется походка, возникает мышечная гипотония, нарушается чувство равновесия, отмечается непроизвольное мочеиспускание и задержка мочи. При тяжёлом течении заболевания возможны судороги, требующие применения антиконвульсантов.
В возрасте 5-10 лет и старше часто развивается синдром запястного канала . Данное состояние требует обязательной коррекции, в противном случае развиваются контрактуры дистальных межфаланговых суставов, пальцы теряют чувствительность, наступает парез мышц большого пальца.
Скопление полисахаридов в глоточном лимфоидном кольце, надгортаннике и трахее является причиной сужения дыхательных путей, развития обструктивных состояний, рецидивирующих инфекций дыхательных путей и среднего уха.
Для больных мукополисахаридозом I типа также характерно поражение глаз в виде прогрессирующего помутнения роговицы, пигментной дегенерации и повышения внутриглазного давления [1] .
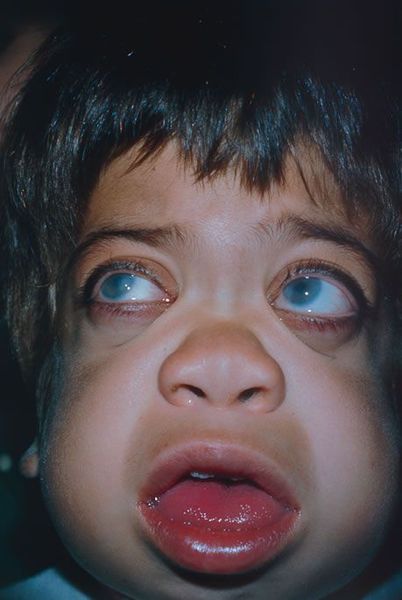
Средняя продолжительность жизни детей с синдромом Гурлер достигает 10 лет. Частой причиной смерти становятся обструктивные заболевания дыхательной системы, острые и хронические инфекционные процессы, патология сердечно-сосудистой системы.
Мукополисахаридоз I H/S (синдром Гурлер — Шейе) является среднетяжёлой формой заболевания. К самым частым симптомам относятся нарушение движения в суставах, снижение прозрачности роговицы и низкий рост [7] .
Первые признаки проявляются к 3-8 годам. Из-за преждевременного сращения черепных швов изменяется форма черепа и нарушается рост головного мозга. Переносица уплощается и западает, губы становятся пухлыми, верхняя челюсть становится меньше, повышается оволосение, кожа утолщается.
Как правило, в течение первых 12 месяцев жизни рост нормальный, но по мере взросления скорости роста снижается и развивается низкорослость. Пропорции тела нарушаются [1] . Суставы становятся тугоподвижными, нарушается форма костей, деформируется грудная клетка, возникает кифоз, сколиоз позвоночника.
С возрастом увеличиваются миндалины, рецидивируют респираторные заболевания, воспаления среднего уха, нарушается проходимость дыхательных путей. Со стороны глаз поражается роговица: происходит её помутнение из-за накопления гликозаминогликанов [7] . Нарушение мозга сопровождается задержкой психического, моторного, лингвистического развития, в исходе развивается деменция. Отмечается гидроцефалия, пахименингит в верхних отделах позвоночника, что приводит к компрессии спинного мозга, развитию патологии в нижележащих отделах [1] . Развивается синдром запястного канала, являющийся причиной контрактур суставов. Со стороны сердечно-сосудистой системы развиваются клапанные аномалии за счёт утолщения створок, нарушение сократимости сердечной мышцы. Увеличение печени и селезенки приводит к диспропорциональному увеличению живота. Патология передней брюшной стенки проявляется пахово-мошоночными грыжами, широким пупочным кольцом с образованием грыж [6] .
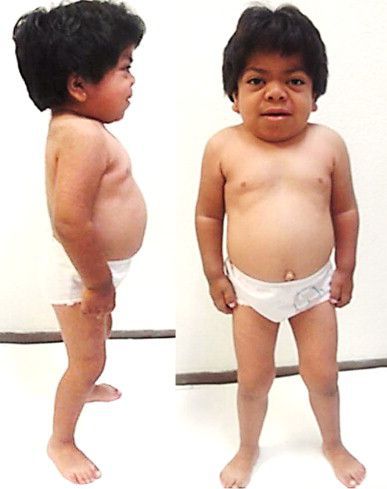
Мукополисахаридоз I S (синдром Шейе, или болезнь Гурлер с поздним началом) — лёгкая форма заболевания. Отличается стёртой клинической картиной. Грубые черты лица, напоминающие гаргулью, также присутствуют, но в более лёгкой степени, чем при синдроме Гурлер. Интеллект, психическое и моторное развитие, как правило, в пределах нормы. Иногда наблюдается незначительное запаздывание развития.
Основными клиническими проявлениями являются поражения суставов, сопровождающиеся нарушением их функции, и отставание в росте [1] . Поражение костей и суставов приводит к тугоподвижности конечностей, болевому синдрому, деформации кистей по типу «лапы с когтями», «пустой стопы», вальгусу в коленных суставах (Х-образным голеням), тоннельный синдром способствует развитию контрактур.
Характерны увеличение печени, селезёнки, образование пахово-мошоночных грыж, слабости пупочного кольца [1] , болезни органов слуха и рецидивирующие инфекционные заболевания органов дыхательной системы, которые приводят к приступам ночного апноэ. Зрение страдает из-за неравномерного помутнения роговицы, повышения внутриглазного давления, пигментной дегенерации сетчатки, что более характерно для больных старше 30 лет.
Часто формируется компрессия срединного нерва, что приводит к развитию карпального туннельного синдрома, связанного со сдавлением срединного нерва между костями, мышцами и связками запястья. Клапанные пороки сердца, расширения аорты могут быть причиной развития сердечной недостаточности.
Мукополисахаридоз II типа (синдром Хантера) бывает тяжёлой и умеренно тяжёлой формы. Такое разделение основано на степени поражения нервной системы и возрасте, в котором проявляются первые признаки болезни.
Синдром Хантера включает в себя множество различных признаков с поражением различных органов и систем [8] . Основу клинической картины составляют нарушения центральной нервной системы, проявляющиеся задержкой умственного развития, огрубление черт лица, низкорослость, нарушение подвижности суставов.
Большинство симптомов схожи с I типом болезни: грубые черты лица, напоминающие гаргулью, увеличенная мозговая часть черепа, выступающий лоб, уплощённая запавшая переносица, плоский нос с укороченными носовыми ходами, выворачивающиеся ноздри, приоткрытый рот, увеличенный язык, толстые губы, низкий рост, укороченная шея, тугоподвижность суставов, повышенное оволосение, пахово-мошоночные и пупочные грыжи. Отличается запоздалым прорезыванием зубов, длинными густыми ресницами, широкими густыми сросшимися бровями [8] . При прогрессировании заболевания волосы осветляются и выпрямляются, становятся сухими и жёсткими.
Для синдрома Хантера характерна сыпь в виде мелких узелков, группирующихся на спине, плечах и бёдрах. Её появление связано со скоплением мукополисахаридов в дерме.
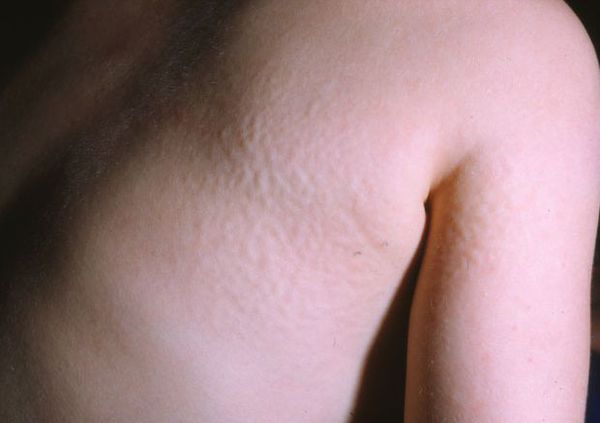
За счёт укорочения и расширения пястных костей, развития пястного тонельного синдрома кисть приобретает когтистую форму. По мере прогрессирования заболевания формируется кифосколиоз, деформируются тазобедренные суставы, возникают различные костные аномалии, увеличивается размер турецкого седла — углубления в клиновидной кости черепа.
Рецидивирующие инфекции органов дыхания и слуха, увеличение миндалин приводят к тугоухости, апноэ во сне. Поражение глаз проявляется помутнением роговицы (реже, чем при других типах болезни), пигментной дегенерацией сетчатки, а при тяжёлых формах происходит дистрофия сетчатки и отёк диска. При развитии внутричерепной гипертензии наблюдается отёк зрительного нерва.
Психическое развитие начинает отставать в возрасте 1,5-3 лет. При тяжёлых формах заболевания к 8-10 годам появляется тяжёлая умственная отсталость и эпилепсия, которая трудно поддаётся лечению. Иногда формируется сообщающаяся гидроцефалия, парезы, потеря чувствительности в конечностях. Известны случаи сдавления спинного мозга, связанные с увеличением толщины оболочек, нестабильностью атлантоаксиального сустава (между затылочной костью и первым шейным позвонком). Это приводит к мышечной слабости, недержанию мочи, задержке мочеиспускания и неуклюжести [6] . Из-за поражения ствола головного мозга нарушается глотание и подвижность нижней челюсти. Возможны псевдобульбарные и бульбарные параличи (связанные с поражением продолговатого мозга).
Большую трудность представляют поведенческие нарушения: гиперактивность, агрессия, упрямство. Часто ухудшают ситуацию проблемы со сном, нарушения слуха. Со временем присоединяются расстройства аутистического спектра. Эти состояния трудно поддаются коррекции. При тяжёлом течении заболевания развивается деменция.
Поражение сердечно-сосудистой системы проявляется клапанными пороками сердца, кардиомиопатией.
С раннего детского возраста отмечается увеличение печени и селезёнки, нарушение переваривания пищи и моторики кишечника. Увеличение языка и поражение височно-нижнечелюстного сустава приводят к нарушению глотания.
Мукополисахаридоз III типа (синдром Санфилиппо) отличается от других тем, что при нём гликозаминогликаны скапливаются по большей части в тканях мозга, а не в соединительной ткани. Самые яркие клинические признаки: задержка психического развития, маловыраженное поражение суставов, лёгкое огрубление черт лица [2] .
Первые проявления возникают на втором году жизни: низкорослость, умеренная тугоподвижность суставов, увеличение печени и селезёнки [9] . До 3 лет дети обычно развиваются в пределах нормы, а затем утрачиваются ранее приобретённые моторные и речевые навыки, нарушается психическое развитие. В дальнейшем возникает нарушение поведения, грубое нарушение психики, переходящее в деменцию. Речь, как правило, не формируется.
Характерны черепно-лицевые изменения: увеличенные лобные бугры, низкие надбровные дуги, широкая спинка носа, густые сросшиеся брови, густые ресницы, сухие и жёсткие волосы, повышенное оволосение [2] .
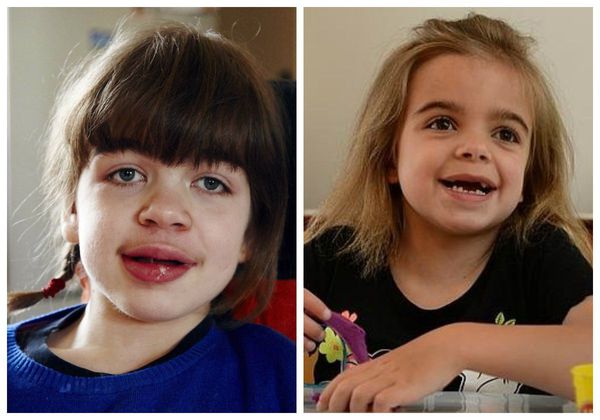
Мукополисахаридоз IV типа (синдром Моркио) сопровождается значительными деформациями костной системы, которые затрагивают в основном руки, ноги и грудную клетку.
В первые месяцы жизни ребёнка признаки заболевания отсутствуют, клиническая картина проявляется только в возрасте 1-3 лет. К 7-8 годам симптомы болезни становятся наиболее яркими. Характерна низкорослость, задержка физического развития. Кожа толстая, малоэластичная. Лицевые признаки: широкий рот, укороченный нос, редкие зубы, дисплазия эмали зубов.
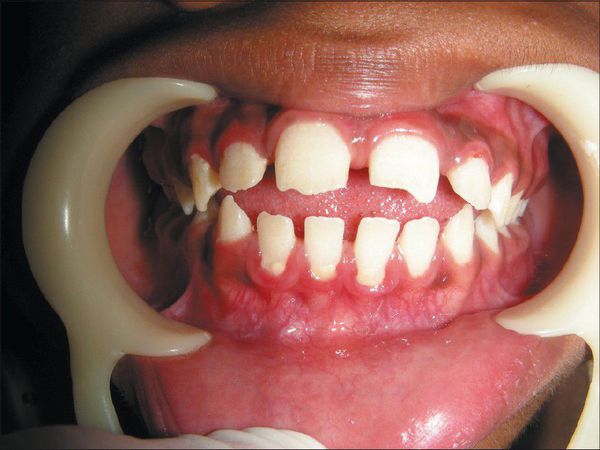
Мышцы гипотоничны, грудная клетка деформирована, отмечается кифосколиоз грудного и поясничного отделов позвоночника. Интеллект не страдает [3] . Суставы тугоподвижны, нередко развиваются контрактуры. Часто появляется шейная миелопатия — поражение волокон спинного мозга с нарушением проведения нервного импульса. Иногда развивается тугоухость. Отмечается слабость апоневроза передней брюшной стенки с образованием грыж [3] .
Мукополисахаридоз VI типа (синдром Марото — Лами) имеет разнообразные проявления, которые прогрессируют с разной скоростью. Характерна низкорослость, снижение зрения, грубые черты лица, тугоухость, снижение подвижности суставов, увеличение печени и селезёнки, поражение сердечно-сосудистой системы и органов дыхания. Интеллект, как правило, в пределах нормы.
Черты лица напоминают гаргулью. Нарушены пропорции тела, формируется карликовость. Суставы деформированы, множественные деформации костей приводят к инвалидизации. Отмечается нарушение развития тел грудных позвонков, их переломы при незначительной нагрузке. Часто возникает сдавление спинного мозга, связанное с нестабильностью шейного отдела позвоночника. Иногда развиваются клапанные пороки сердца, приводящие с сердечной недостаточности. Со стороны желудочно-кишечной системы наблюдается синдром раздражённого кишечника, увеличение печени и селезёнки.
Мукополисахаридоз VII типа (синдром Слая) проявляется увеличением печени и селезёнки, образованием пахово-мошоночных или пупочных грыж, низкорослостью, деформацией грудной клетки, кифосколиозом в поясничном и крестцовом отделах позвоночника, искривлением нижних конечностей, рецидивирующими респираторными заболеваниями, грубыми чертами лица с широко расставленными глазами, уплощённой переносицей и вывернутыми вперёд ноздрями. Иногда наблюдаются клапанные пороки сердца и кардиомиопатия.
Осложнения мукополисахаридоза
Основными осложнениями течения мукополисахаридозов различных типов являются тяжёлые рецидивирующие респираторные инфекции (риниты, синуситы, отиты, ОРЗ), приводящие к дыхательной недостаточности, патология сердечно-сосудистой системы и поражение головного мозга.
Сам мукополисахаридоз не приводит к смерти. Больные чаще всего умирают в результате сердечной и дыхательной недостаточности, которые развиваются на фоне заболевания [5] .
Диагностика мукополисахаридоза
Диагностика основана на определении характерных клинических (внешних) признаков и изучении активности гликозаминогликанов в крови. Последнее исследование проводится следующим образом: на специальные бланки с фильтровальной бумагой капается капиллярная кровь пациента, затем высушивается и отправляется в лабораторию, где проводится ферментный анализ. Возможно определение концентрации гликозаминогликанов в моче, но часто встречаются ложные результаты [4] [14] .
Также в рамках диагностики проводится генетическое исследование. Оно заключается в поиске мутаций в определённых генах, отвечающих за развитие мукополисахаридозов.
Дополнительная диагностика заключается в поиске поражений органов и систем с помощью различных исследований:
- УЗИ — выявляет увеличение печени и селезёнки, пороки развития сердца и увеличение его размеров;
- рентгенография — выявляет патологию костей и суставов;
- электрокардиография — выявляет кардиомиопатию, патологию сократимости и проводимости;
- электромиография — позволяет диагностировать нарушения в проведении возбуждения по нервным волокнам к мышцам;
- аудиометрия — помогает выявить проблемы со слухом [5][14] .
При проведении рентгенологических исследований в случае мукополисахаридоза I H типа выставляется диагноз «множественный дизостоз» — нарушения развития костей. Диафизы длинных трубчатых костей расширены, при рентгенологических исследованиях отмечается изменение структуры метафизов и эпифизов. Ключицы короткие и толстые, рёбра приобретают форму весла, части, расположенные ближе к позвоночнику, сужены, а передние — толстые и широкие [1] . Фаланговые кости рук и ног укорочены, имеют форму трапеции, диафизы широкие.
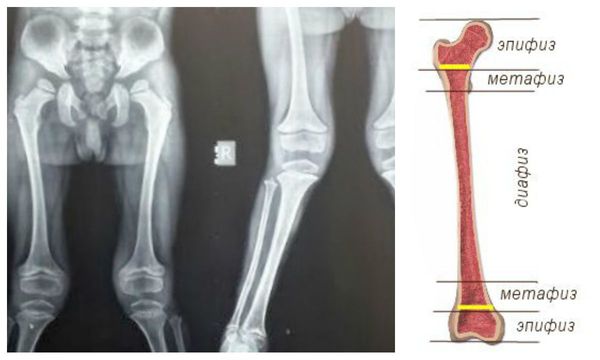
Формируются уплощение позвонков, сколиоз, кифосколиоз. Позвонки в поперечнике широкие, но они низкие. В деформированных участках отмечается недоразвитие поперечных отростков либо их «языкообразная» трансформация.
До рождения мукополисахаридоз и другие хромосомные нарушения можно обнаружить с помощью амниоцентеза (исследования околоплодной жидкости) или биопсии хориона (слоя плаценты). Также риск появления мукополисахаридоза у младенца можно определить ещё до зачатия. Для этого родителям нужно пройти генетический анализ [14] .
Лечение мукополисахаридоза
Симптоматическое лечение заключается в устранении грыжи, удалении миндалин и ортопедической хирургии. Проводится коррекция искривлений позвоночного столба при помощи транспедикулярной фиксации металлическими конструкциями, исправление деформаций костей и суставов, коррекция грудной клетки. Это позволяет облегчить болевой синдром, уменьшить риск сдавления спинного мозга. Также проводится шунтирование желудочков головного мозга при гидроцефалии.
Патогенетическое лечение, направленное на причину болезни, возможно при помощи трансплантации костного мозга и стволовых гемопоэтических клеток и заместительной ферментной терапии [5] . Трансплантация возможна от родственного донора либо из стволовых клеток пуповинной крови родственного донора после проведения химиотерапевтической или лучевой супрессии. Данный вид лечения не получил широкого распространения из-за высокого риска осложнений (инвалидизации или смерти). Заместительная ферментная терапия способна остановить прогрессирование заболевания, частично улучшить уже сформированные патологии. Применение такой терапии позволяет добиться регресса заболевания и существенно улучшить прогноз жизни.
В настоящее время ферментная терапия разработана только для некоторых типов мукополисахаридозов. При I типе используется альдуразим . Он представляет собой рекомбинантную человеческую альфа-L-идуронидазу. Препарат вводится внутривенно каждую неделю в течение 4-х часов [4] . Для мукополисахаридоха II типа показана элапраза [2] .
Мукополисахаридозы относятся к редким заболеваниям, поэтому препараты для их лечения выделяются на государственном уровне и предоставляются пациентам в рамках оказания бесплатной медицинской помощи [5] . В некоторых случаях заместительная терапия является пожизненной [14] .
Прогноз. Профилактика
При отсутствии должного лечения прогноз неблагоприятный, так как больные погибают от осложнений. Практически всегда заболевание приводит к инвалидизации. Однако всё зависит от типа заболевания. В некоторых случаях пациенты могут жить так же долго, как и здоровые люди [14] .
Профилактика заключается в генетическом консультировании пар, вступающих в брак, особенно при отягощённом семейном анамнезе.
Возможно проведение преимплантационной генетической диагностики в циклах ЭКО у пар с высоким риском рождения ребёнка с мукополисахаридозом либо генетическая диагностика на ранних сроках беременности с возможностью прерывания беременности при выявлении заболевания у плода.
Перспективным является создание генетических паспортов, выбор партнёра с отсутствием соответствующих мутаций.
Список литературы
- Министерство здравоохранения РФ. Федеральные клинические рекомендации по диагностике и лечению мукополисахаридоза I типа. — 2013.
- Союз педиатров России. Клинические рекомендации по ведению детей с мукополиса-харидозом III типа. — 2013.
- Союз педиатров России. Федеральные клинические рекомендации по оказанию мади-цинской помощи детям с мукополисахаридозом IV типа. — 2013.
- Министерство здравоохранения РФ. Федеральные клинические рекомендации по ока-занию медицинской помощи детям с мукополисахаридозом I типа. — 2015.
- Министерство здравоохранения РФ. Федеральные клинические рекомендации по диагностике и лечению мукополисахаридоза II типа. — 2013.
- Hamosh A. Hurler syndrome. Mucopolysaccharidosis type IH; MPS1-H // Online Mendelian Inheritance in Man. — 2018
- Przylepa K. A. Scheie syndrome. Mucopolysaccharidosis type IS; MPS1-S. Mu-copolysaccharidosis type V, Formerly, MPS V, Formerly; MPS5, Formerly // Online Mendelian Inheritance in Man. — 2002
- Hamosh A. Mucopolysaccharidosis, type II; MPS2 // Online Mendelian Inher-itance in Man. — 2018.
- Hamosh A. Mucopolysaccharidosis, type IIIA; MPS3A // Online Mendelian In-heritance in Man. — 2018.
- Hamosh A. Mucopolysaccharidosis, type IIIB; MPS3B // Online Mendelian In-heritance in Man. — 2012.
- Kniffin C. L. Mucopolysaccharidosis, type IIIC; MPS3C // Online Mendelian Inheritance in Man. — 2011.
- O’Neill M. J. F. Mucopolysaccharidosis, type IIID; MPS3D // Online Mendelian Inheritance in Man. — 2010.
- Боровикова О. И. Типы мукополисахаридозов и клинические проявления // Счастливая беременность. — 2019.
- Demczko M. Mucopolysaccharidoses // MSD Manuals. — 2018.
Источник https://www.krasotaimedicina.ru/diseases/traumatology/mucopolysaccharidosis
Источник https://probolezny.ru/mukopolisaharidoz/