Синдром Марфана

Синдром Марфана — наследственное заболевание, которое проявляется системным поражением соединительной ткани в организме человека. В результате болезни происходят нарушения строения скелета и кожи, работы глаз, сердечно-сосудистой, дыхательной и других систем организма. Эту генетическую мутацию нельзя предотвратить или вылечить, но правильно подобранное лечение способно продлить пациентам жизнь и предупредить развитие опасных осложнений.
Причины синдрома Марфана
Данное генетическое заболевание вызвано дефектом гена FBN1 в длинном плече 15 хромосомы. Этот ген кодирует белок гликопротеин фибриллин-1, который отвечает за прочность и эластичность соединительной ткани. Соответственно, все проявления патологии связаны с тем, что соединительнотканные структуры в организме человека теряют свои нормальные свойства.
Наследуется мутация по аутосомно-доминантному признаку, то есть дети получают патологический ген от родителей, которые страдают от патологии. При этом шанс ребенка получить мутацию от одного из родителей составляет 50% (рис. 1). Синдром не передается через поколение: здоровые дети больных родителей не могут передать ген своим потомкам.
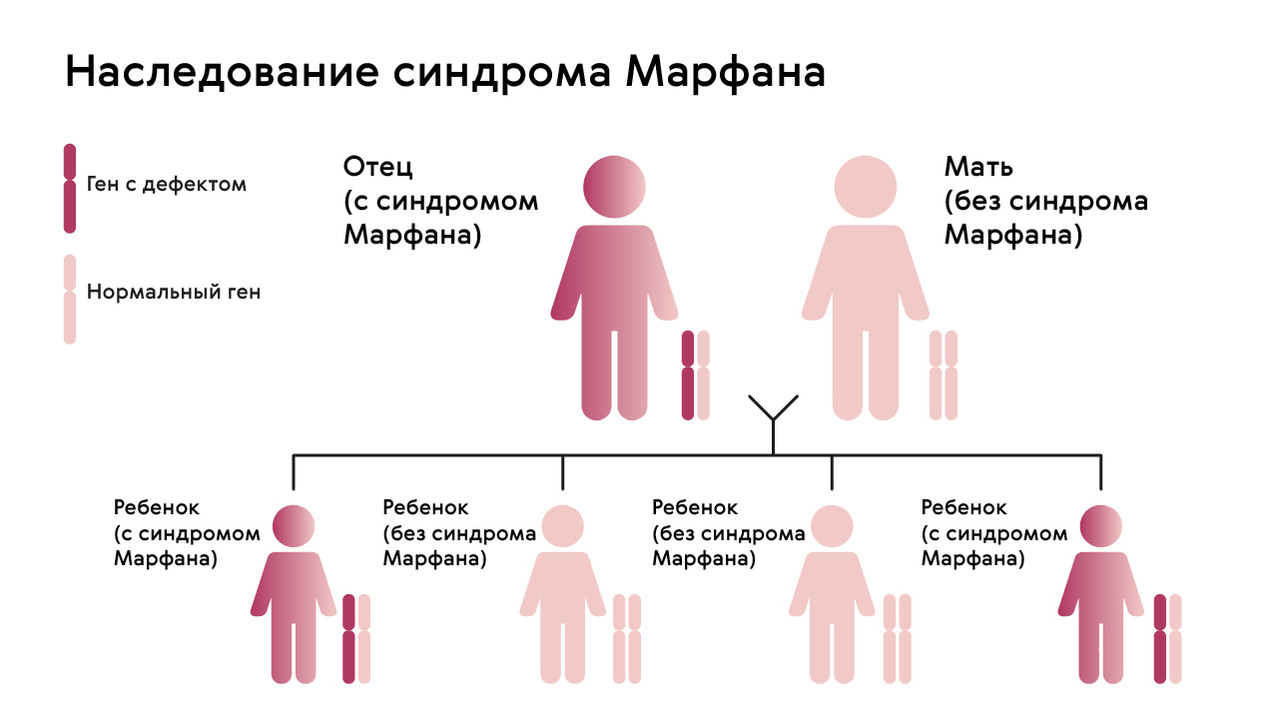
Однако примерно у 25% людей с синдромом Марфана никто из родителей не оказывается носителем аномалии гена FBN1: в таком случае мутация развивается спонтанно.
До сих пор не выявлено определенных факторов риска развития этого генетического нарушения: заболевание встречается одинаково часто среди мужчин и женщин, а его распространенность не зависит от расы или этнической группы. Частота заболеваемости у этой патологии составляет примерно 1 случай на 5–10 тысяч.
Если клинические признаки мутации ярко выражены, заподозрить болезнь можно уже в первые месяцы жизни ребенка, но стертые формы заболевания часто проявляются уже во взрослом возрасте, когда пациент обращается к врачам по поводу различных проявлений синдрома.
Важно! Не стоит записываться на генетическое обследование в качестве медосмотра. Поиски «поломки» гена FBN1 оправданы только в случае, если болезнь проявляет себя характерными признаками: бессимптомное носительство этой мутации невозможно. Если у одного из родителей установлен этот диагноз, будущей маме следует пройти генетическое обследование еще до родов. Это позволит заранее узнать, передалась ли аномалия ребенку.
Классификация синдрома Марфана
Выделяют несколько форм заболевания в зависимости от особенностей клинических проявлений генетической мутации.
Существуют две основные клинические формы патологии:
- Стертая. Таким пациентам «везет» больше: аномалия у них проявляется поражениями только одной-двух систем организма, а симптомы выражены незначительно. Люди могут жить практически нормальной жизнью, несмотря на болезнь.
- Выраженная. В таких случаях поражаются три и более систем организма, либо значительно нарушается функционирование одной из систем.
В зависимости от степени проявления выделяют легкие, среднетяжелые и тяжелые формы синдрома Марфана. Тяжелые патологии встречаются гораздо реже: частота их выявления составляет примерно 1 на 25–50 тысяч человек.
Принципиальную роль в определении прогноза болезни играет характер ее течения:
- Прогрессирующий. В этом случае постоянно появляются новые симптомы заболевания, степень тяжести увеличивается, а с каждым годом жизни пациента возрастают риски фатальных осложнений.
- Стабильный. Такой характер считается наиболее благоприятным: у пациентов со стабильными проявлениями синдрома Марфана клиническая картина практически не меняется на протяжении жизни.
Выделяют три разных, но похожих заболевания:
- Синдром Марфана — стертая форма патологии с положительным результатом генетического тестирования.
- Болезнь Марфана — классическая клиническая картина с подтвержденным семейным наследованием.
- Марфаноподобный синдром — проявление патологии соединительной ткани без генетической мутации.
Первые признаки заболевания чаще всего проявляются еще в детском возрасте. К подростковому периоду становится понятно, насколько быстро у пациента прогрессирует болезнь, вызванная мутацией гена FBN1.
Симптомы синдрома Марфана
Проявления генетического дефекта могут быть выражены в разной степени: от легкого изменения строения соединительной ткани до тяжелых нарушений жизненно важных функций организма. Иными словами, внешние признаки аномалии у разных пациентов могут значительно отличаться, несмотря на одинаковый генетический дефект.
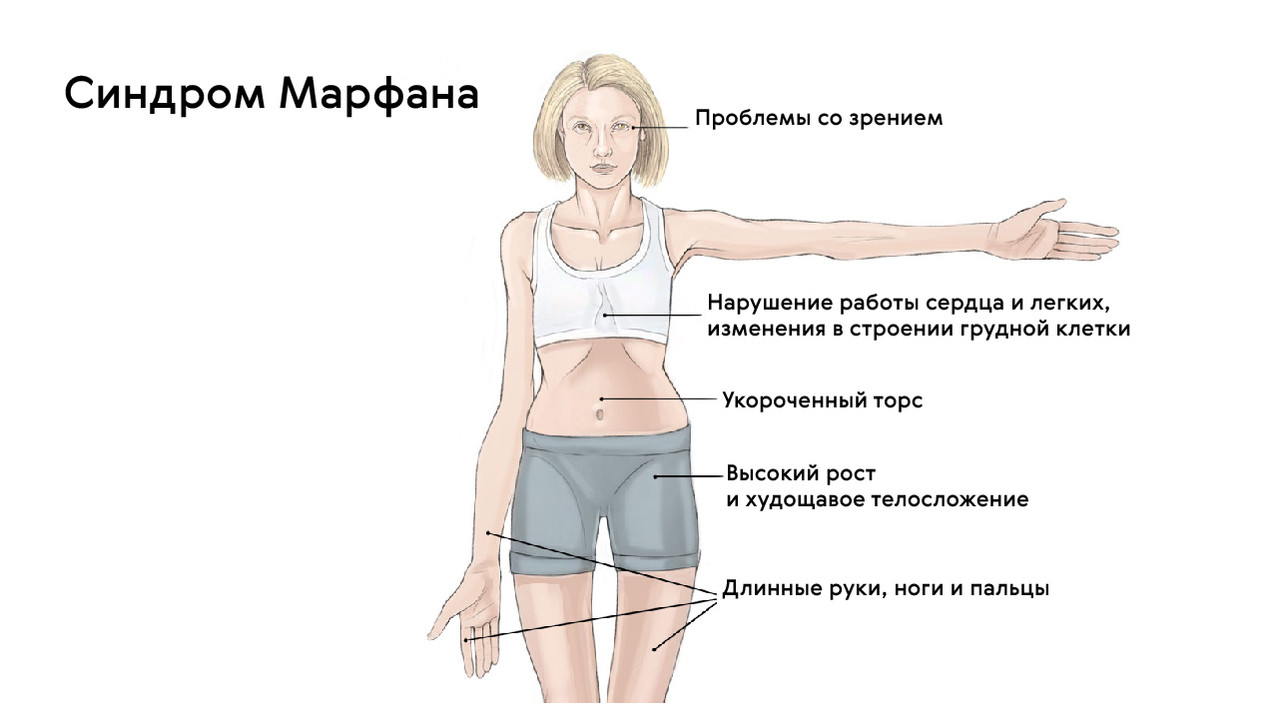
Классической триадой синдрома Марфана считаются: скелетные нарушения, смещение хрусталика и расслоение аорты (рис. 2). Также системное поражение соединительной ткани у пациентов становится причиной развития нарушений работы практически всех органов и систем организма.
Костно-мышечная система
Выраженность симптомов поражения опорно-двигательного аппарата зависит от тяжести случая и особенностей организма пациента.
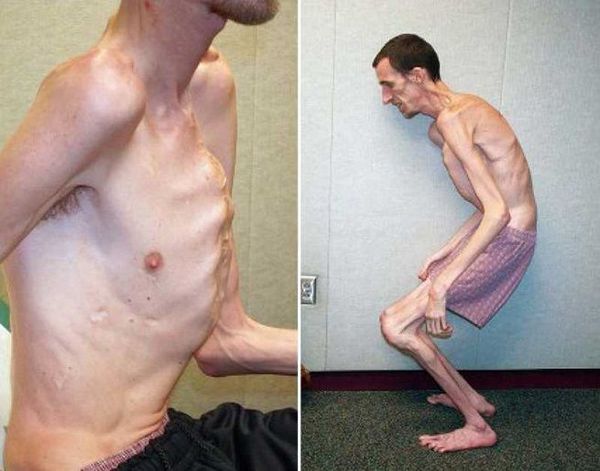
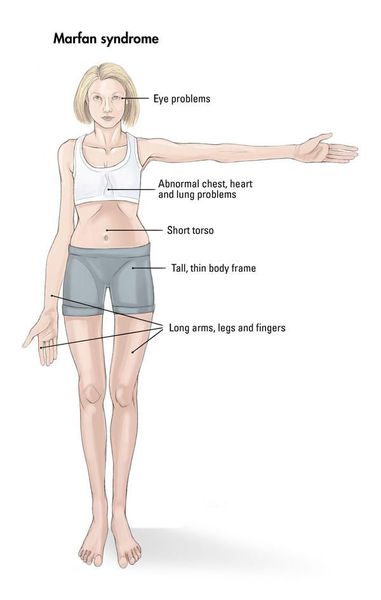
Для людей с синдромом Марфана характерен чрезвычайно высокий рост: обычно дети «перерастают» всех членов семьи. При этом часто, особенно в детском возрасте, привлекает внимание нестандартная длина рук: их размах оказывается больше, чем длина тела.
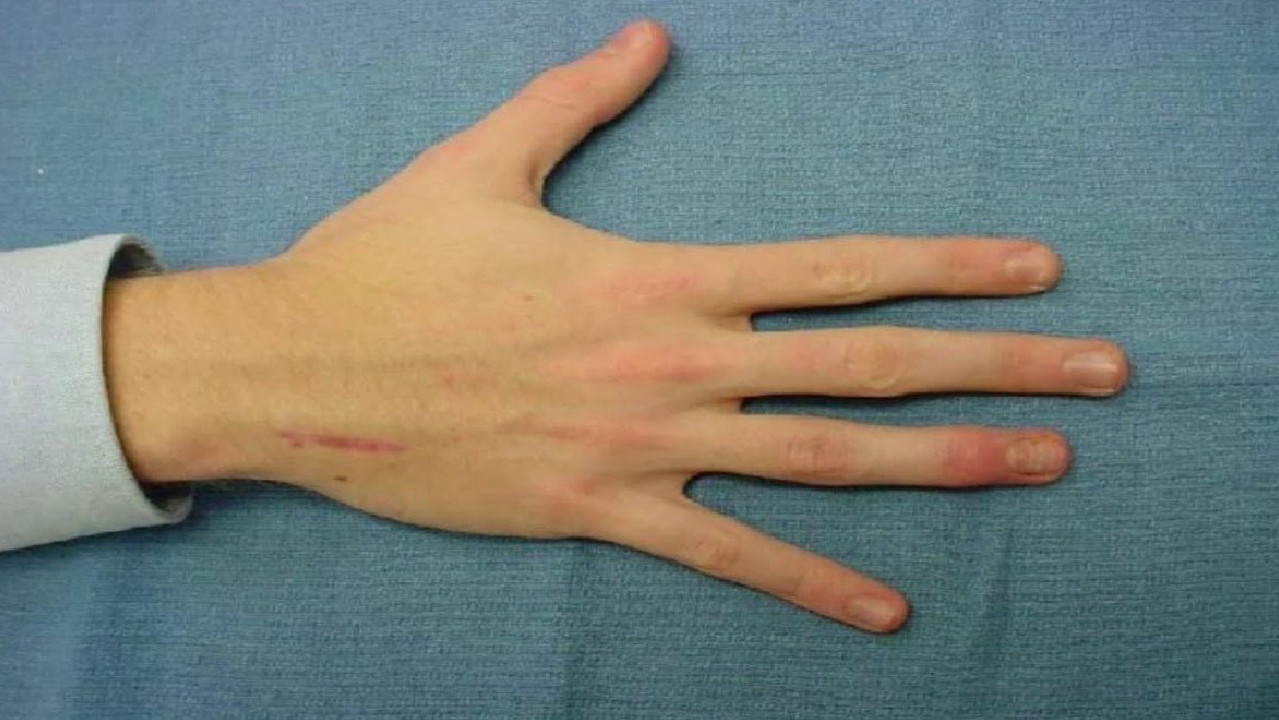
Яркий симптом болезни — патологически удлиненные и тонкие пальцы, так называемые «пальцы паука» (арахнодактилия) (рис. 3).
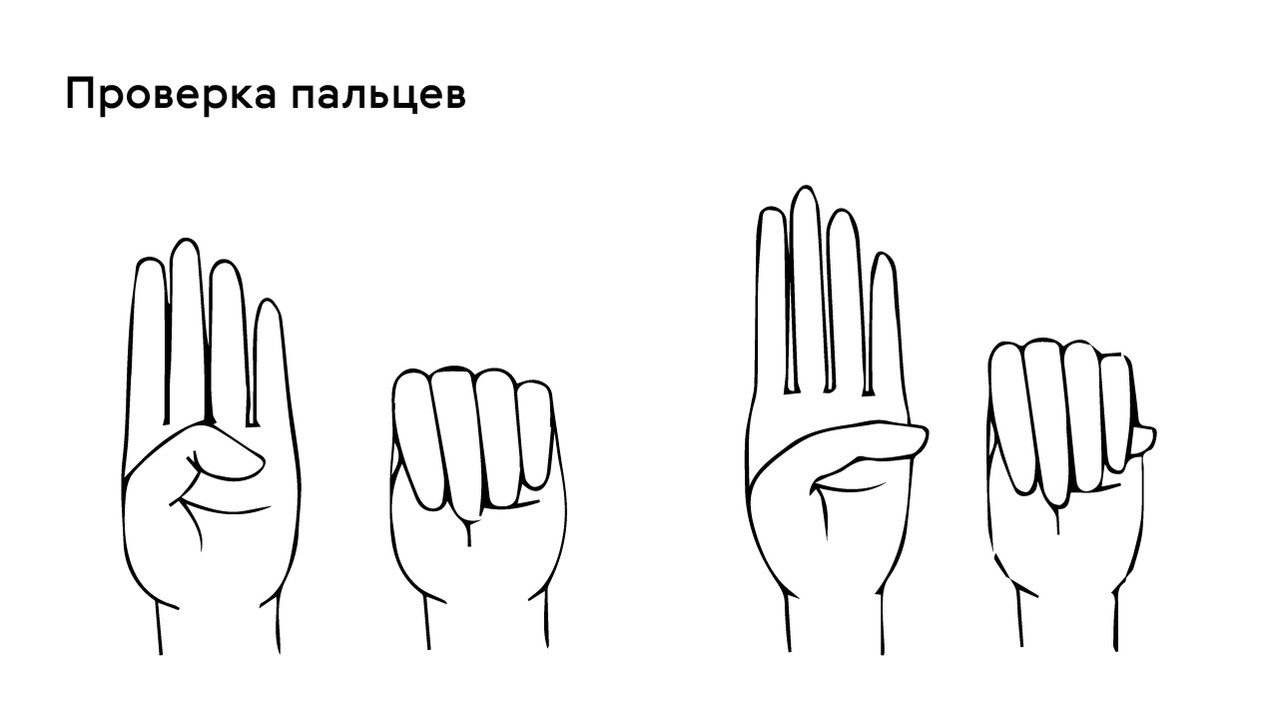
Проверить наличие симптома можно с помощью теста большого пальца кисти — у пациентов с арахнодактилией часть большого пальца (дистальная фаланга) выступает за край сжатой в кулак ладони (рис. 4).
Лицо людей с синдромом Марфана обычно вытянутое и худое. Этому способствует высокое положение свода верхнего неба, удлиненный череп и патологическая худоба.
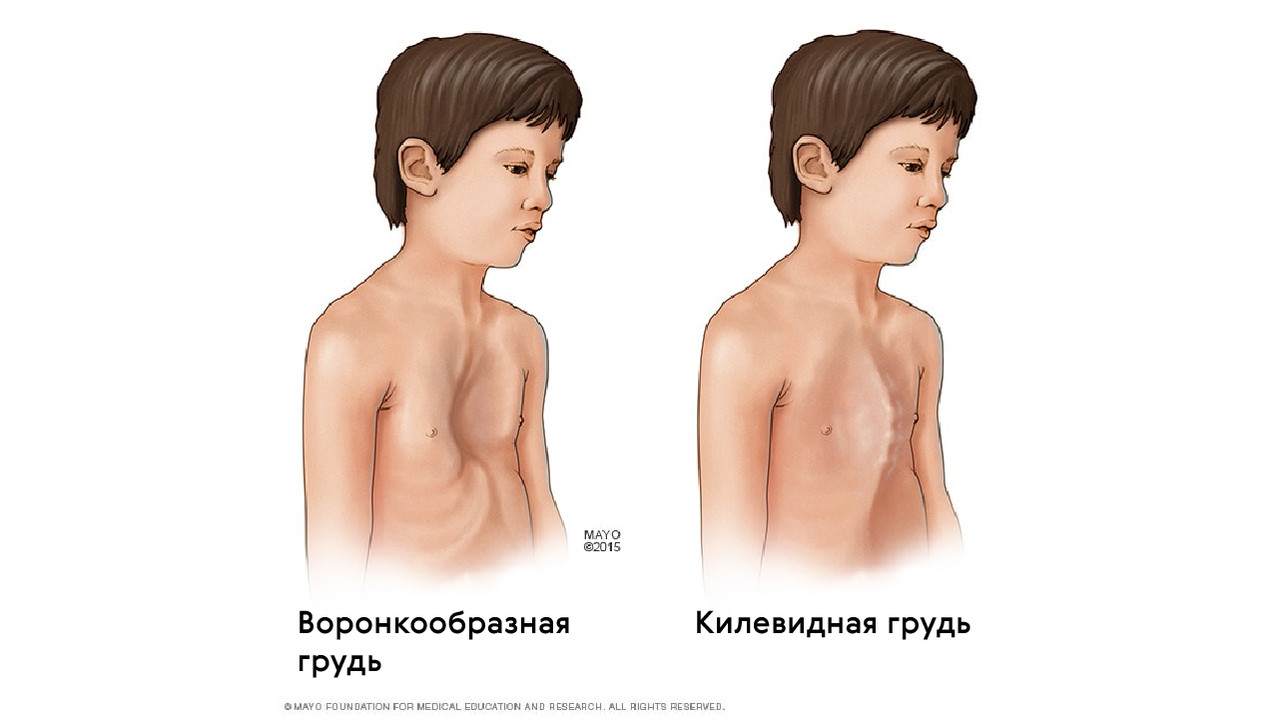
Также для таких пациентов характерны деформации грудной клетки, которые могут быть в двух вариантах: смещение грудины внутрь (воронкообразная грудь) или наружу (килевидная грудь, рис. 5).
Осанка пациентов с синдромом Марфана в большинстве случаев нарушена. Чаще всего определяются различные степени выраженности сколиоза (отклонение позвоночного столба в сторону) или кифоза (формирование «горба»).
Кроме того, пациенты с FBN1 мутацией часто страдают от:
- плоскостопия;
- повышенной подвижности всех суставов;
- слабости связочного аппарата.
У пациентов с синдромом Марфана часто плохо развиты мышечные структуры и практически нет подкожно-жирового слоя. Движения пациентов с этой патологией неловкие, они часто получают различные травмы.
Кожа
Высокий темп роста и нарушения выработки белков соединительной ткани определяют патологии кожи у людей с мутацией гена FBN1. Клинически это проявляется в виде повышенной растяжимости кожных структур с образованием светлых полос — «растяжек» (стрий).
Зрение
Дефекты гена FBN1 определяют склонность к патологиям зрительной системы. Чаще всего повреждения глаз у пациентов с синдромом Марфана включают в себя:
- выраженную близорукость;
- подвывих или изменение положения хрусталика;
- высокий риск внезапной отслойки сетчатки глаза.
Кроме того, у таких пациентов гораздо раньше может развиться катаракта или глаукома: те патологии органа зрения, которые считаются возрастными у здоровых людей.
Органы дыхания
В легких пациентов с синдромом Марфана может патологически разрастаться соединительная ткань. Это приводит к формированию сужения бронхов и легочного фиброза. Нередко на фоне генетической мутации развивается бронхиальная астма или хроническое обструктивное заболевание легких. Генетическая аномалия также определяет возможность развития спонтанного пневмоторакса — неотложной ситуации, в которой в полость вокруг легких попадает воздух, и легкое резко уменьшается в размерах («спадается»).
Желудочно-кишечный тракт
Процессы пищеварения у людей с FBN1 мутацией меняются: нарушается моторика кишечника, появляются патологии желчного пузыря, часто развиваются гастриты, язвенные дефекты, дисбиоз.
Почечный аппарат
У пациентов с синдромом Марфана чаще находят аномалии почек: опущение органов, расширение почечных лоханок, патологическую подвижность почек.
Нервная система и психическая сфера
Хотя в большинстве случаев у пациентов с синдромом Марфана не происходит нарушений работы мозговых структур, некоторые патологические изменения нервной системы могут присутствовать. Например, расширение соединительнотканной капсулы, которая окружает спинной мозг, может приводить к нарушениям движений в нижних конечностях, работы мочевого пузыря и кишечника. Для таких пациентов характерно развитие синдрома хронической усталости — астения, склонность к депрессии. Интеллектуальная деятельность в большинстве случаев не нарушена, даже наоборот: среди пациентов с синдромом Марфана есть люди с интеллектом значительно выше среднего.
Сердечно-сосудистая система
Кардиологи выявляют нарушения ритма сердца у людей с синдромом Марфана. У пациентов с этой патологией часто нарушается структура аортального клапана — соединительнотканной перегородки, которая предупреждает обратный ток крови из аорты в сердце. Это приводит к развитию порока сердца — аортальной недостаточности. Также могут развиваться другие пороки сердца, например, пролапс или недостаточность митрального клапана, а на пораженных участках часто развивается инфекционно-воспалительный процесс — бактериальный эндокардит.
Самую большую опасность представляют патологические изменения в главном сосуде организма — аорте. У 65–100% людей с синдромом Марфана есть большой риск поражения луковицы (наиболее близкая к сердцу часть аорты) и восходящей дуги этой артерии — тех частей, которые непосредственно выходят из сердца. Поскольку внутренний слой стенки сосудов также содержит волокна соединительной ткани, они склонны к износу, а давление крови в аорте выше, чем в других участках сосудистого русла. Это приводит к тому, что сосуд постепенно расширяется, и может произойти патологическое скопление крови между сосудистыми стенками с формированием мешковидного выпячивания (аневризмы) или спонтанный разрыв артерии.
Почему при определении признаков синдрома Марфана нужно обратиться к врачу?
Сама по себе генетическая аномалия совместима с жизнью. Однако опасны последствия болезни, вызванной FBN1 мутацией:
- разрывы крупных сосудов, чаще всего — аорты;
- хроническая сердечная недостаточность — неспособность сердца обеспечивать необходимую работу для кровоснабжения всех органов;
- снижение остроты зрения или полная потеря зрительной функции.
Разрыв аневризмы аорты или другого магистрального сосуда часто заканчивается моментальным летальным исходом. Хроническая сердечная недостаточность может перейти в острую форму, а без экстренной медицинской помощи также привести к фатальным последствиям — внезапной коронарной смерти. Именно эти осложнения чаще всего приводит к гибели детей с синдромом Марфана. Особая опасность ждет женщину с синдромом мутации гена FBN1 во время беременности: повышенная нагрузка на аорту в разы увеличивает риск ее разрыва.
Чтобы предупредить развитие опасных осложнений и компенсировать возникающие нарушения, родителям нужно как можно раньше обратиться за медицинской помощью при первом подозрении на синдром Марфана у ребенка. При этом важно не только однократно провести обследование, но и стать на учет к врачам, которые занимаются коррекцией проявлений синдрома:
- специалисту по генетическим болезням;
- кардиологу;
- ортопеду-вертебрологу;
- дерматологу;
- офтальмологу;
- гастроэнтерологу.
Список специалистов зависит от степени выраженности заболевания, при этом регулярно необходимо проходить комплексные профилактические осмотры для раннего выявления новых нарушений.
Синдром Марфана — болезнь гениев?
С синдромом Марфана связаны не только многочисленные поводы для обращения к врачам. Часто люди с мутацией гена FBN1 компенсируют физические проявления болезни интеллектуальными способностями, поэтому это генетическое заболевание даже называют «синдромом гениев». Считается, что повышенный выброс адреналина из-за патологических изменений в надпочечниках определяет высокий тонус умственной и психической активности у таких пациентов. Именно поэтому в числе людей с синдромом Марфана можно найти известных личностей. Например, Юлию Цезарю, Аврааму Линкольну и Шарлю де Голлю патология не помешала стать известными политическими деятелями; Ганс Христиан Андерсен и Корней Чуковский создали уникальные литературные произведения, а Никколо Паганини прославился как гениальный музыкант.
Современные знаменитости также не скрывают свои недостатки и становятся еще более популярными из-за генетического дефекта. Например, солисту американской рок-группы Deerhunter Брэдфорду Коксу нетипичная внешность придает особый шарм, а испанский актер Хавьер Ботет очень востребован, поскольку правдоподобно и талантливо играет отрицательных героев в голливудских фильмах ужасов (рис. 6).

Диагностика синдрома Марфана
Диагностика генетической аномалии включает в себя комплекс мероприятий по определению всех симптомов болезни, а также изучению вероятности развития мутации:
- Сбор жалоб — детальное изучение всех патологических признаков.
- Определение анамнеза — выяснение состояния здоровья родителей.
- Тщательный осмотр, измерение роста, размаха рук и других антропометрических показателей. Скрининговый тест для детей в возрасте 7–18 лет — это измерение длины среднего пальца руки. У пациентов с синдромом Марфана показатель превышает отметку в 10 см.
Генетическое обследование включает в себя выявление генотипа ДНК — идентификацию мутаций в гене FBN1. При возможности назначают специфические лабораторные тесты — определение выведения с мочой метаболитов соединительной ткани, таких как оксипролин и гликозаминогликаны.
Чтобы подтвердить нарушения развития соединительной ткани и оценить степень выраженности мутации гена FBN1, пациентам с подозрением на синдром Марфана назначают:
- ЭКГ;
- УЗИ сердца;
- КТ-ангиографию аорты и других сосудов;
- КТ грудной и брюшной полостей;
- МРТ позвоночника и головного мозга;
- специфические обследования на осмотре у офтальмолога;
- биопсию кожи.
Для окончательного определения диагноза используют общепринятые Гентские критерии 2010 года, согласно которым диагноз устанавливают в случаях:
- подтвержденной мутации гена FBN1 и расширения корня аорты или эктопией хрусталика;
- подтвержденного расширения корня аорты в сочетании с эктопией хрусталика;
- подтвержденной эктопии хрусталика в сочетании с любыми признаками системного поражения соединительной ткани.
Важно! Существует группа «марфаноподобных» синдромов, при которых внешне пациенты очень напоминают больных с аномалией гена FBN1, но причина их патологии скрывается в других нарушениях. К примеру, гомоцистинурия — это обменное заболевание, которое проявляется системными изменениями соединительной ткани, но может приводить к внезапным инсультам и существенно замедляет умственное развитие ребенка. Поэтому важно точно определить причину заболевания соединительной ткани и своевременно начать лечение.
Лечение синдрома Марфана
К сожалению, на сегодняшний день лекарственные методы терапии этой генетической патологии еще не разработаны. Однако пациентам с синдромом Марфана важно соблюдать все назначения врачей, чтобы устранить симптомы патологии и замедлить темпы ее развития.
Лечение зависит от клинических проявлений болезни:
- при аневризме аорты назначают препараты, которые снижают частоту и силу сердечных сокращений, снимая избыточную нагрузку на сосуды;
- пациентам с синдромом Марфана часто назначают антигипертензивные препараты для снижения артериального давления;
- хондроитин и глюкозамин относятся к естественным компонентам соединительной ткани — их прием улучшает структуру хрящей и предупреждает патологии суставов;
- для стимуляции образования коллагена выписывают специальные БАДы — L-карнитин, витамины из групп С, D, Е, В, а также кальций, цинк и другие пищевые добавки.
Пациентам противопоказаны физические нагрузки, постоянная активность, травмоопасные игры. Рацион питания людей с синдромом Марфана должен быть насыщен белками, полезными жирными кислотами, микро- и макроэлементами. Для поддержки структур скелета пациентам с мутацией в гене FBN1 показано ношение корсетов, укрепление мышц с помощью ЛФК и оздоровительного массажа.
В некоторых случаях может помочь только хирургическое лечение — операции по замене части аорты, клапанов, исправлению костных патологий или коррекции патологий глаза, которые существенно снижают риски опасных осложнений.
Прогноз
Современные методы исследования в медицине позволяют выявлять заболевание у детей в раннем возрасте. Это помогает повысить качество жизни таких пациентов и предупредить раннюю смертность. Продолжительность жизни людей с синдромом Марфана при бережном отношении к своему здоровью достигает 70 лет. Прогноз болезни во многом зависит от выраженности сердечно-сосудистых патологий, поскольку выживание пациентов с этой генетической аномалией определяет состояние аорты и риск ее спонтанного разрыва. Такие люди требуют постоянного наблюдения у врачей различных специальностей для своевременной коррекции проявлений синдрома.
Заключение
Конечно, жизнь с этой генетической мутацией становится сложнее, но при правильном подходе к собственному здоровью и своевременному обследованию у врачей пациентам с синдромом Марфана удается компенсировать все проявления заболевания и не допустить развития фатальных осложнений.
Активисты с синдромом Марфана создают тематические сообщества по всему миру: мощная поддержка людей с такой же генетической аномалией позволяет пациентам не чувствовать себя одинокими.
Источники
- Отделение синдрома Марфана. Андалузский институт высокотехнологичной кардиологии (Бенальмадена и Фуэнхирола, Малага, Испания).
- Dadkhah, E., Ziaee, M., Davari, M. H., Kazemi, T., & Abbaszadegan, M. R. (2012). Informative STR Markers for Marfan Syndrome in Birjand, Iran. Iranian journal of basic medical sciences, 15(5), 1020–1025.
- Прийма, Н. Ф., Попов, В. В., Комолкин, И. А., & Афанасьев, А. П. (2013). Аневризма аорты у пациента с синдромом Марфана. Педиатр, 4 (1), 100-108.
- Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, Hilhorst-Hofstee Y, Jondeau G, Faivre L, Milewicz DM, Pyeritz RE, Sponseller PD, Wordsworth P, De Paepe AM. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010 Jul;47(7):476-85. doi: 10.1136/jmg.2009.072785. PMID: 20591885.
Синдром Марфана — симптомы и лечение
Что такое синдром Марфана? Причины возникновения, диагностику и методы лечения разберем в статье доктора Боровиковой Ольги Игоревны, генетика со стажем в 8 лет.
Над статьей доктора Боровиковой Ольги Игоревны работали литературный редактор Маргарита Тихонова , научный редактор Сергей Федосов

Генетик Cтаж — 8 лет
«Evi Clinic» («Эви Клиник»)
Клиника «OXY-center»
Медицинский центр «Андромед»
Дата публикации 9 апреля 2018 Обновлено 12 декабря 2021
Определение болезни. Причины заболевания
Синдром Марфана (Marfan; СМ) — генетически обусловленное заболевание, при котором происходит системное поражение соединительной ткани. [1]
Причины синдрома Марфана
Этиологией заболевания является мутация в гене FBN1 (фибриллина 1), расположенном в коротком плече пятнадцатой хромосомы в локусе 21.1. [2]
Тип наследования синдрома — аутосомно-доминантный. Для болезни характерна высокая пенетрантность (частота появления гена) и различная экспрессивность. [5]
Соотношение представителей мужского пола и женского одинаковое.
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением — это опасно для вашего здоровья!
Симптомы синдрома Марфана
Наблюдается постоянно прогрессирующее развитие заболевания. У новорожденных детей выявляются удлинённые тонкие пальцы на верхних и нижних конечностях и удлинённые тонкие конечности (долихостеномелия). [1] У таких пациентов, помимо долихостеномелии, отмечается:
- повышенное физическое развитие;
- недостаток веса;
- удлинённый череп;
- вытянутое лицо;
- арахнодактилия (аномально удлинённые узкие пальцы);
- слабость и недоразвитие мышечной системы и жировой клетчатки;
- неловкие движения. [3]
Кожа имеет повышенную растяжимость, разболтанные суставы. У большинства больных наблюдается высокое аркообразное нёбо, изменения формы грудной клетки (воронкообразная, килевидная) и искривления позвоночника (сколиоз в 60%, кифоз (изгиб позвоночника с образованием горба), ювенильный остеохондроз), уплощение свода стопы, аускультативные признаки порока сердца (шумы). [4] Длина третьего пальца руки — 10 см и больше (скрининговый тест у детей 7-18 лет): возрастает соотношение размаха верхних конечностей к длине тела.
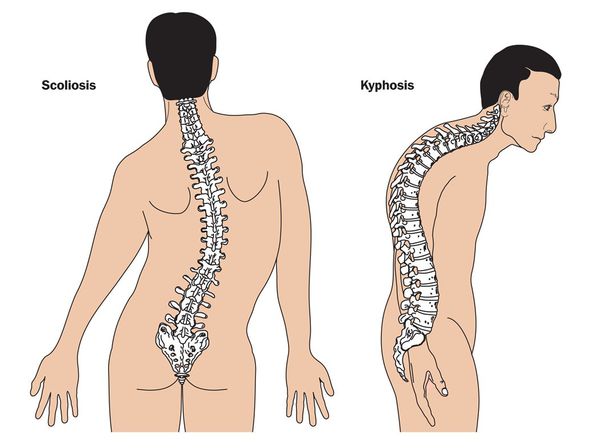
Офтальмологические симптомы (близорукость, подвывих хрусталика в 75% случаев, его округлость или гипоплазия, отслойка сетчатки) и астенические признаки (усталость, вялость) обращают на себя внимание со второго года жизни, изменения формы грудной клетки появляются в возрасте старше четырёх лет, патология сердца и сосудов выявляется в дошкольном возрасте. [1]
Почти у всех больных выявляются пороки сердца и аорты. Часты бедренные и паховые грыжи, поражение клапанов в венах, их варикозное расширение, геморрагический синдром, рецидивирующие вывихи, поражение лёгочной системы (самопроизвольный пневмоторакс, эмфизематозное расширение лёгких), опущение почек. [2]
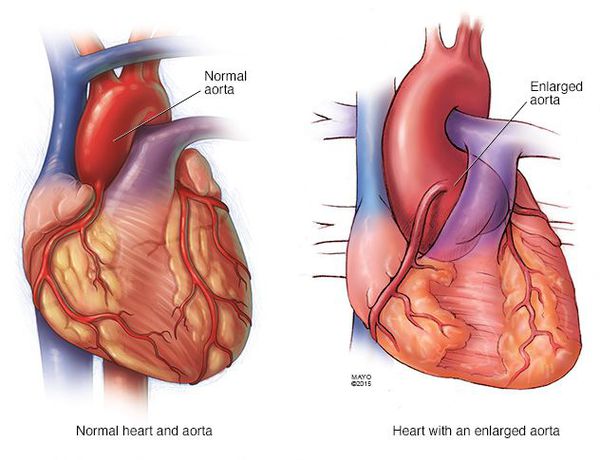
В четверти случаев зарегистрировано снижение интеллекта, у половины пациентов выявляются нарушения эмоционально-волевой сферы. Часто появляются депрессивные состояния, нейроциркуляторная дистония. [3]
По данным многих исследований, абсолютное большинство больных с синдромом Марфана отмечают ухудшение эмоционального фона, утрату чувства радости и увлечённости профессиональной деятельностью, частую смену настроения, повышенную возбудимость, чувство тревоги. Результатом этого является снижение социальной активности, ухудшение качества жизни и значительное уменьшение социальной адаптации. [3]
У таких пациентов часто наблюдается трахеобронхиальная дискинезия (нарушение дыхательной системы) за счёт слабости соединительнотканного каркаса бронхов. Это проявляется рецидивирующими воспалительными заболеваниями бронхолегочной системы, обструктивными нарушениями, бронхиальной астмой, эмфиземой лёгких (повышенное содержание воздуха в лёгочной ткани). [4] Встречаются осложнения, которые проявляются скоплением воздуха в грудной клетке, сопровождающиеся сдавлением лёгких и средостения (срединной области грудной клетки), подкожной эмфиземой. Наблюдается неадекватный ответ на бронхолитики. Обструктивные явления (непроходимость) затрагивают преимущественно верхние отделы респираторного тракта. [3]
Описаны характерные изменения на электрокардиограмме, включающие синдром раннего возбуждения желудочков, преждевременные желудочковые комплексы, нестабильность конечной части желудочкового комплекса в задненижних отведениях. [3]
Патология ритма чаще всего проявляются блокадой правой ножки пучка Гиса или смешанной экстрасистолией. [6]
У больных синдромом Марфана с патологией ритма сердечной деятельности и проводимости синдром вегетативной дисфункции чаще протекает по ваготоническому типу, в виде пресинкопальных, обморочных и астеновегетативных состояний, болезненных ощущений в области сердца, цефалгии напряжения (головной боли) и зачастую сочетается с психопатологическими расстройствами. [4]
Органы пищеварения также задействованы в патологическом процессе, что проявляется дискинезией (нарушением моторики) билиарного тракта со снижением моторики гладкомышечной мускулатуры, недостаточностью кардии, грыжевыми выпячиваниями пищеводного отверстия диафрагмы, аномалиями желчевыводящих протоков, долихосигмой (увеличением сигмовидной кишки), хроническим гастродуоденитом (воспалением слизистой желудка и двенадцатиперстной кишки), дисбиозом (нарушением нормальной микрофлоры) кишечника, изменениями поджелудочной железы. [3]
У пациентов с синдромом Марфана чаще, чем у здоровых людей, встречаются приобретённые аномалии почек: повышенная подвижность почек, нефроптоз (опущение почки), пиелоэктазии (аномальное расширение лоханок), повышена частота удвоения почек.
Патогенез синдрома Марфана
Более половины веса человека представлено соединительной тканью, из неё состоит наша главная опора — скелет, внешние покровы — кожа. Сосуды, кровь и лимфа тоже состоят из соединительной ткани.
К клеткам соединительной ткани относятся фибробласты и их разновидности (остеобласты, хондроциты, одонтобласты, кератобласты), макрофаги (гистиоциты) и тучные клетки (лаброциты). [7]
Мезенхима — проводник конституциональных, генетических и эпигенетических составляющих жизни человека. Патология соединительной ткани детерминирует определенное патологическое действие на весь организм в целом, на его физиологию и его конституциональные особенности. [3]
При болезни Марфана происходит замена нуклеотидов в гене, содержащем информацию о структуре пептида фибриллина-1. Этот белок относится к гликопротеидам, принимает участие в микрофибриллярном комплексе, он обеспечивает основу эластических фибрилл соединительной ткани.
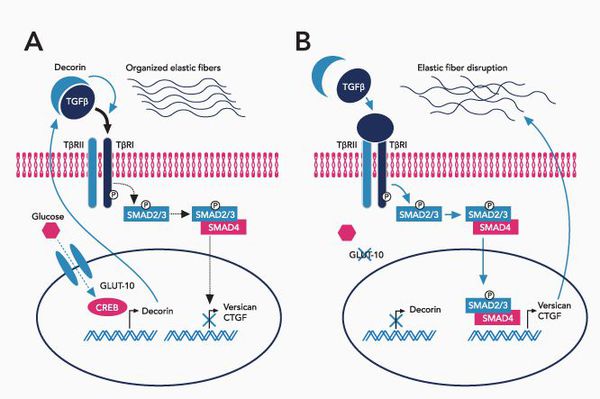
Межклеточный матрикс позволяет соединительной ткани поддерживать постоянную структуру, в нём находится огромное количество факторов роста, которые обеспечивают постоянное обновление клеток.
В крупных сосудах, связочном аппарате содержится большое количество эластиновых фибрилл, поражение которых и даёт основные клинические проявления синдрома Марфана.
При синдроме Марфана значительно поражается трансформирующий фактор роста бета (TGF-β), нарушается связывание его неактивной формы, что приводит к повышению биоактивности данного фактора, с чем связано появление многих проявлений болезни. [4]
Патология фибриллина приводит к патологии формирования волокон, что вызывает утерю прочности и эластичности кожи и других соединительнотканных структур.
Изменение структуры коллагеновых волокон приводит к нарушению первичного звена гемостаза у пациентов с синдромом Марфана. [6]
Имеются данные о дефектах мембранных и цитоплазматических механизмов проведения сигнала непосредственно в самом тромбоците, приводящих к нарушениям агрегации (объединения). Показано наличие самостоятельного мембранного дефекта тромбоцитов, протекающего с нарушением реакций высвобождения и транспорта внутриклеточного кальция. [6]
Эластические фибриллы имеют вполне определенные механизмы участия в системе гемостаза. В сосудах с низкой скоростью сдвига происходит адгезия («прилипание») тромбоцитов к эластину через фибронектин. [7] Регистрируется снижение его уровня в крови у людей с синдромом Марфана. Фибронектин, в свою очередь, образуется в клетках эндотелия и участвует в последующих репаративных процессах, создавая основу для производства других компонентов соединительной ткани — фибробластов. [4] Таким образом, совершенно неоспоримо участие сосудистой стенки в реакциях свертываемости крови, и неизбежен вывод о возможных патологиях протекания нормальных гемостатических процессов при изменении состояния её структурных компонентов и процессов сосудистой регуляции.
Отмечена роль гормонального дисбаланса в развитии и усугублении дефектов соединительнотканных структур. [3]
Тромботические проявления детерминированы нарушением реологии (вязкости) крови в патологически извитых сосудах брахиоцефальной зоны. [3]
Поражение желудочно-кишечного тракта детерминировано тем, что эта система богата коллагеном. Наблюдаются дискинезия билиарного тракта по гипомоторному типу, грыжи пищеводного отверстия диафрагмы, аномалии желчных путей, долихосигма, хронический гастродуоденит со стёртой клинической картиной, склонностью к торпидному течению. [3]
Классификация и стадии развития синдрома Марфана
Код синдрома Марфана по Международной классификации болезней (МКБ-10): Q87.4.
- стёртая (поражено не более двух систем, изменения выражены незначительно);
- выраженная (незначительные изменения в трёх системах либо значительное поражение одной и более систем).
Выделяют различные типы по степени тяжести:
Частота тяжёлых форм — 1 к 25000-50000 (при общей частоте диагностированных случаев 1 к 10000-15000).
По характеру течения:
- прогрессирующая форма;
- стабильная форма.
Чаще всего первые признаки синдрома Марфана проявляются еще в детском периоде, с возрастом происходит прогрессирование симптомов, усиление клинических проявлений.
Осложнения синдрома Марфана
К самым частым осложнениям синдрома Марфана относятся:
- Снижение зрения, вплоть до слепоты, обусловленное слабостью цинновой связки (ресничного пояска) и подвывихом, вывихом хрусталика. [7]
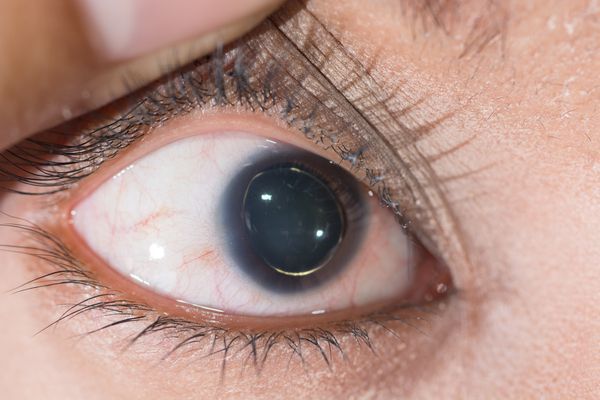
- Сердечная недостаточность по застойному типу, обусловленная нарушением сократимости сердечной мышцы, недостаточностью митрального клапана. [6]
- Разрывы крупных сосудов, связанные с дилатацией (расширением), истончением стенки сосудов. Чаще всего происходит поражение аорты (в основном из-за изменения гемодинамики при беременности). [7]
- Расслаивающая аневризма аорты, приводящая к смерти больных.
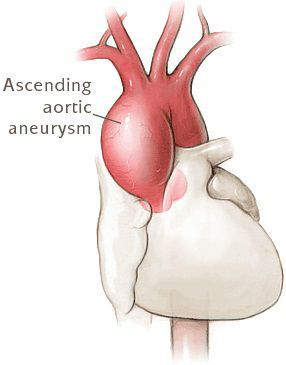
Диагностика синдрома Марфана
Диагностика Синдрома Марфана основывается на клинических данных, выявлении изменений в гене FBN1. [5]
Часто при сборе генеалогического анамнеза выявляются родственные случаи со скрытым течением заболевания. [1]
Способы обнаружения арахнодактилии: [3]
- Симптом Steinberg (признак первого пальца). Первый палец виден из-под hypothenar при напряжённом кулаке.
- Симптом Walker-Murdoch (признак запястья). При обхватывании кистью в области лучезапястного сочленения контралатеральной верхней конечности первый палец заходит за пятый.
- Определение пястного индекса. Определяется при помощи рентгенографии. Средняя длина пясти, делённая на усреднённую ширину отрезка от второй до четвертой пястной кости. При нормальном соотношении этот показатель соответствует 5,4-7,9, в то время, как при синдроме Марфана — больше 8,4.
В 2010 году группа специалистов систематизировала международные Гентские критерии для верификации синдрома Марфана. Верификация зависит от данных генеалогического анамнеза. [3]
При отсутствии генеалогического анамнеза:
- увеличение диаметра аорты >, = 2 ϭ + эктопия хрусталика = СМ;
- увеличение диаметра аорты >, = 2 ϭ + выявленные изменения в гене FBN1 = CM;
- увеличение диаметра аорты >, = 2 ϭ + >, = 7 системных признаков = СМ;
- эктопия хрусталика + наличие изменений в гене FBN1 + дилатация аорты = СМ;
При наличии генеалогического анамнеза:
- Эктопия хрусталика + случай СМ в семье = СМ;
- >, = 7 системных проявлений + случай СМ в семье = СМ;
- увеличение диаметра аорты >, = 2 ϭ + случай СМ в семье = СМ.
В пятнадцати процентах появление ребёнка с синдромом Марфана спорадическое (случайное), у родителей могут быть слабые проявления. У родственников пациентов встречаются заболевания желудочно-кишечного тракта, поражения позвоночника, заболевания глаз. [3]
При малейшем подозрении на синдром Марфана необходима консультация офтальмолога. В анализе мочи таких пациентов отмечается повышение уровня оксипролина, гликозаминогликанов, но эти показатели низкоспецифичны, могут быть при различных дисплазиях соединительной ткани. Выделение оксипролина является показателем тяжести заболевания. Наблюдается нарушение свертываемости крови на тромбоцитарном уровне. [3]
Оценка системных признаков вовлечённости соединительной ткани
Источник https://medportal.ru/enc/rheumatology/systemic/sindrom-marfana/
Источник https://probolezny.ru/sindrom-marfana/